How to Python
Jump to navigation
Jump to search
| Previous: Dictionaries | Next: Programme |
Required course material for the lesson
Powerpoint: How to python
Video: Common beginner mistakes
Subjects covered
Various mistakes and bad ideas, not well thought through sentence constructs.
Avoid hardcoding.
Exercises to be handed in
- Calculate the the standard deviation (1.8355) of the numbers in ex1.dat. The formula leads to directly to a two-pass algorithm (looking at all the numbers twice), where you will have store all the numbers in memory in order to calculate SD. The inspired programmer will find a one-pass algorithm, where you calulate SD just by looking at a number once, thereby not using significant memory. The genius will explain why there is a difference between the two results.
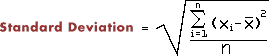
- In the files geneA.txt, geneB.txt, all the way down to geneE.txt you have normalized mRNA expression data taken at the time of discovery of colon cancer for a number of patients and their survival. This is basically 2 columns in each file; The mRNA expression (x) and the number of months (y) the patient survived. For each gene you have to make a simple linear regression analysis and find 3 numbers; the α (the intercept - where the line cuts the Y-axis) and β (the slope) coefficient that describes the line running through the data points best, and the correlation coefficient (r) which describes the fitness of the line. You must identify the gene that best indicates how long the patient survives. For every gene you start calculating these values.
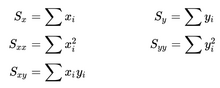 n = number of observations.
n = number of observations.
From the values you can compute the required parameters.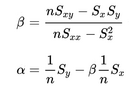

Remember to say which gene best describes survival - and why. A survival prediction can be made by calculating β * x + α, given x which is the mRNA expression.
Note: The genes will in reality interact with each other in ways that totally destroys our basic assumption for making a linear regression: That the data (gene expressions) are independent.
Make your code in a general way - there can for example be more data files. Make it easy to add them. - Repeat the previous exercise again with a new type of data file gene_combined.txt, which is more typical in real life. All genes are in one tab separated file. There are 3 columns; gene name, normalized mRNA expression and survival in months. There is no particular order in which the data appears and data lines for several genes might be mixed within each other.
Again, make general code. There can be more or fewer genes, and you do not need to know there names beforehand.




Exercises for extra practice
- Compute the LIX score of a text. Ask for a file name of a text file. You have to create the file yourself, maybe the abstract of an article or a fairy tale. Count the number of words, the number of long words (more than 6 letters) and the number of sentences in the text. Compute the lix score as described on the wikipedia page.
- This exercise is loosely described. You need to consider it before starting on it.
As we know, DNA is described with the letters ATCG. This just 4 letters from the alphabet. What if we decided to use more of the alphabet to describe DNA. For example B could replace AA, D replace AT, and so forth. Basically, we are using other letters to describe 2 bases instead of 1. Why do that? Well, we will save storage space. We will effectively cut down the space used for sequence to half. The exercise is then to invent such a conversion scheme, read the dna7.fsa file, convert the sequence DNA to the new scheme and save the sequences in converted.fsa.
Consider the following, but do not take on too much at the same time:
1) Make a program similar to this that converts back again to real DNA sequence.
2) What to do if the sequence does not have an even number of bases - there would be one base left after conversion.
3) DNA is not always perfectly sequenced, sometimes we have a non-ATCG in the sequence. The IUPAC codes tells what letters we could expect.
4) The range of chars we can use for the conversion is large, see the ASCII table of chars to use. If we made codes for 3 bases instead of 2 bases, we could save 2/3 of the space instead of just half.
5) IRL, we have compression programs like zip or gzip which will compress our sequences better than anything we can do here. It is just a practice exercise :-)