Before you start: please install the FigTree viewer on your computer.
In this exercise you will analyze the evolutionary relationship between HIV-related viruses
from humans (HIV1 and HIV2), and monkeys (SIV). (For more information about AIDS, HIV1, HIV2, and
SIV, see this explanation)
- Open the sequence file (polseqs.fasta), select
the entire file, and copy the sequences.
- Align the Pol sequences in using the mafft server at EBI with default settings as follows:
First paste the sequences into the input window on the linked mafft page, then click the
"submit" button.
Once the alignment is done, save the resulting alignment as a fasta file as follows: right-click the "Download alignment file" button on the mafft output page, and then save the file using "Save linked file as" (or whatever it is called in your particular browser). Make sure you can find the file again!
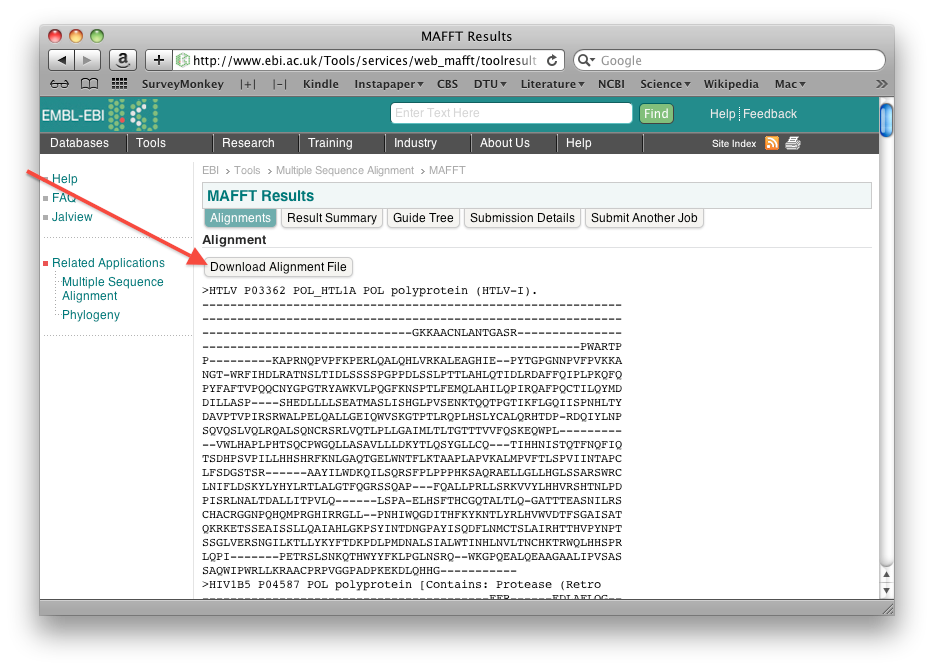
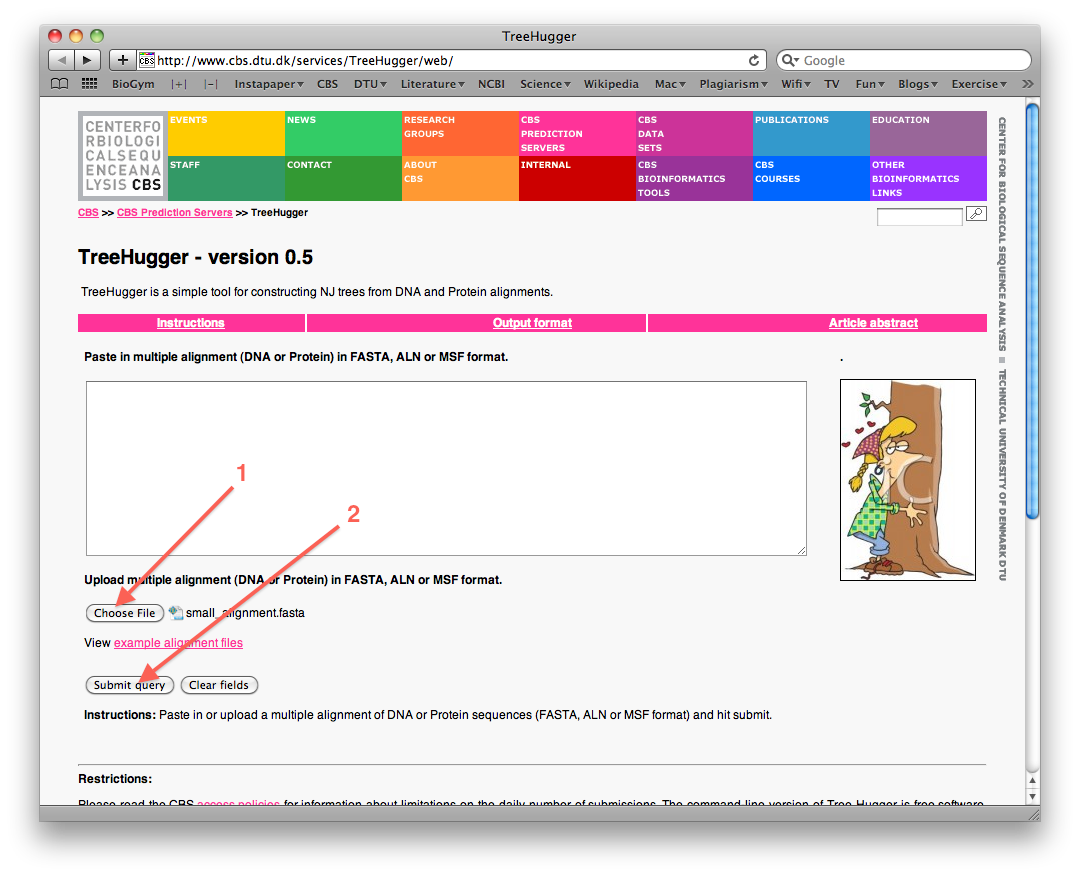
- Open the TreeHugger web server. (The TreeHugger server constructs a neighbor joining tree from an aligned set of sequences).
- Select the option to upload a file (see figure below), then choose the Pol-protein alignment file you just saved on your harddisk, and finally click "Submit Query" to construct the neighbor joining tree:
- When the run is done, right-click the "Download data in Newick/Phylip format" link to save the tree file as a text file on your harddisk (again make sure you can find it later). You will notice that the treefile is in the parenthesis-based format we discussed previously in the lecture:
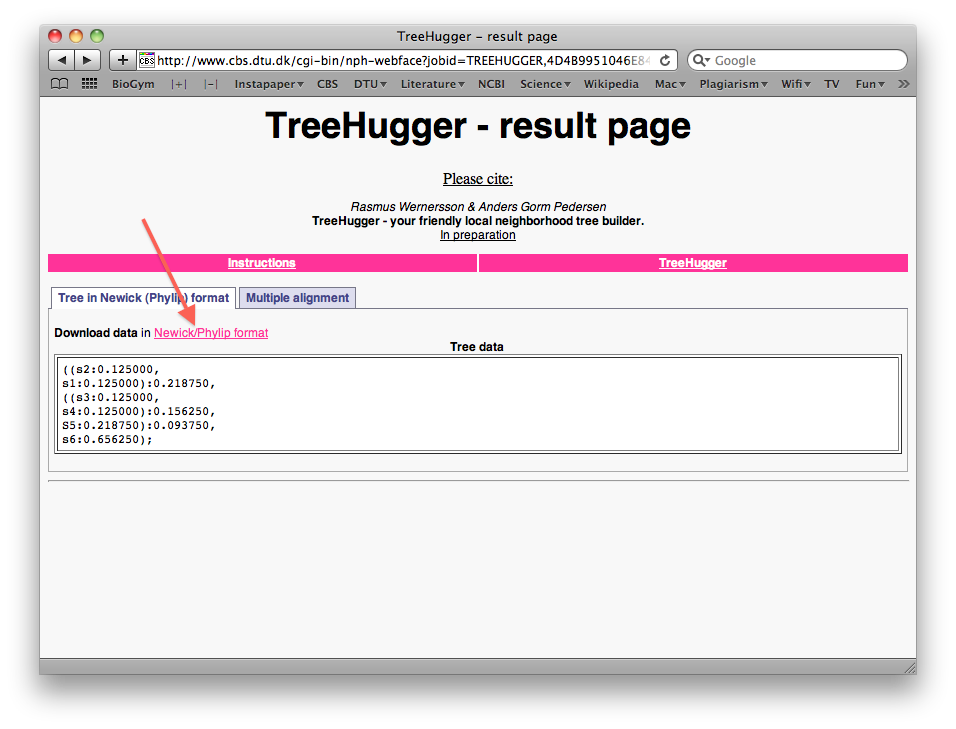
- Open the FigTree treeviewer that you have previously installed on your own computer and use File->Open to open the treefile you just saved.
- The view that you will see first is presumably a rooted view similar to the one below. However, it is important to realize that we have not explicitly rooted the tree yet, so the root in this view has been chosen randomly. A more realistic view can be seen by clicking the unrooted view button (see figures below):
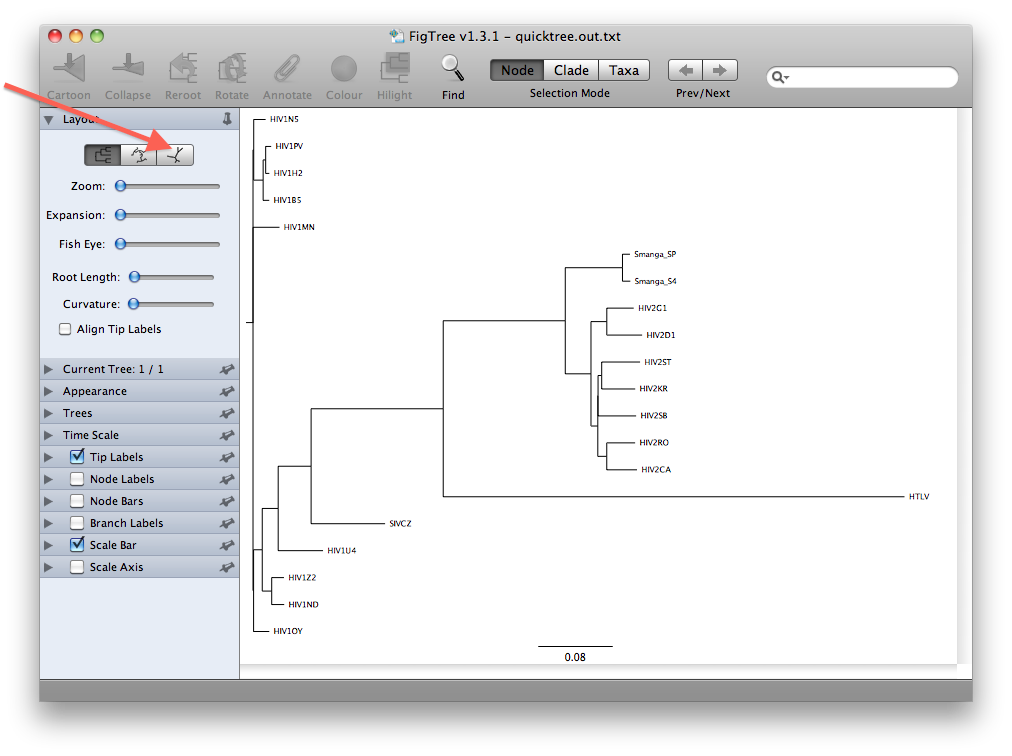

- The last figure above shows the unrooted tree. For now, however, go back to the (pseudo)rooted view you started out with. We wil now place the root by using the HTLV Pol sequence as a so-called outgroup. Click the branch leading to the HTLV sequence such that it gets selected (see figure below). Then click the "Reroot" button, which will subsequently root the tree on the selected outgroup:
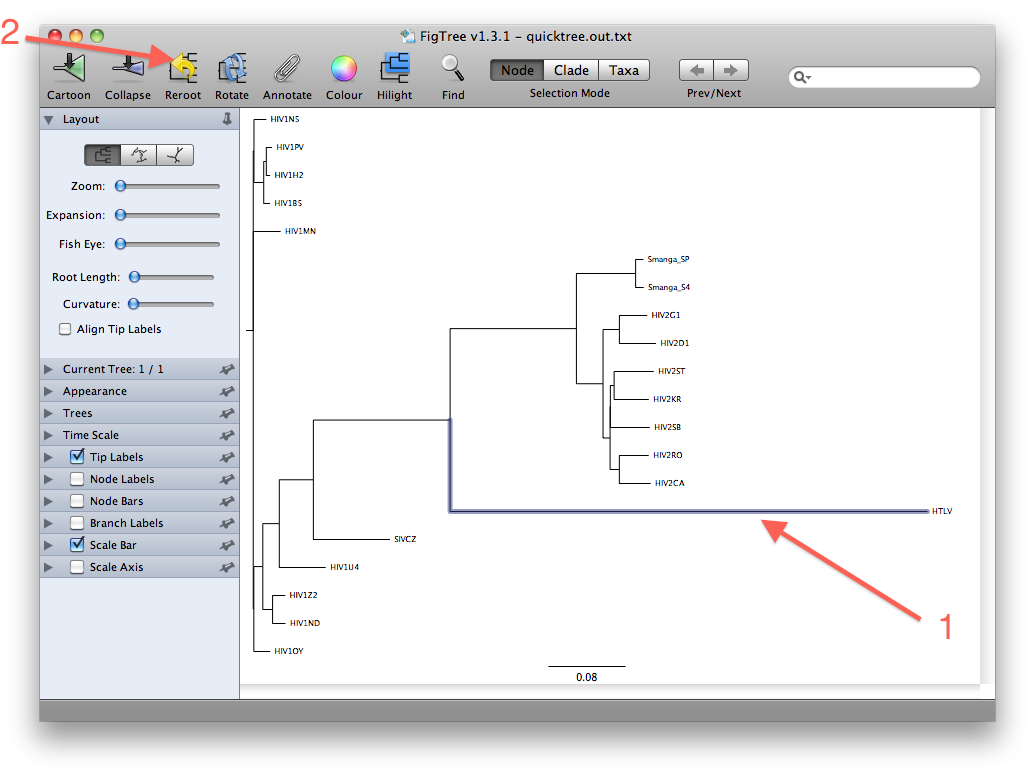
The rationale for using an outgroup to place the root of the tree is as follows: our data set consists of sequences from HIV-1, HIV-2, SIV and HTLV. We know from other evidence that the lineage leading to HTLV branched off before any of the remaining viruses diverged from each other. The root of the tree connecting the organisms investigated here, must therefore be located between the HTLV sequence (the "outgroup") and the rest (the "ingroup"). This way of finding a root is called "outgroup rooting".
- Inspect the rooted tree that you get as a result of rerooting and consider what this tells you about the origin of HIV viruses.
When you have pondered the problem for a while you can read this short explanation that I have prepared: Origin of HIV1 and HIV2.