Dict techniques
| Previous: Set techniques | Next: Regular expressions |
Required course material for the lesson
Powerpoint: Dictionaries
Resource: Clean Code Every time you read it, you will take something from it.
Resource: Example code - Dicts
Video: Live Coding
Subjects covered
- Dictionaries - dicts - which are unordered tables of data.
- Dict methods and functions
- Dict tips and tricks
- Dict algorithms
Exercises to be handed in
- Create a dictionary where the keys are codons and the value are the one-letter-code for the amino acids. The dictionary will function as a look-up table. You can find a codon list here. You are meant to make the dict "by hand" as there is a structure lesson in that. Add a bit of smart code that tests if you made the dict right.
Extra: If you feel like it you can in addition make a program that constructs the dict from a file, which you are responsible for making. - Use the dictionary from the previous exercise and your previous functions fastaread() and fastawrite() in a program, that translates all the nucleotide fasta entries in dna7.fsa to amino acid sequence. Save the results in a file aa7.fsa in fasta format. Since the sequences are now consisting of amino acids add 'Amino Acid Sequence' to each header. The STOP codon is NOT a part of the amino acid sequence. Think about what STOP means.
- In the file ex5.acc are a lot of accession numbers, where some are duplicates. Earlier we just removed the duplicates, but now we should count them. Make a program that reads the file once, and writes a file order5.acc with the unique accession numbers and the number of occurrences in the file. A line should look like this: "AC24677 2", if this accession occurs twice in ex5.acc. The accession numbers must be written in order, which means the accession with most duplicates is on top (the beginning) and the least on bottom. If two accessions have the same amount of duplicates, they need to be ordered according to the accession name, i.e. AC543322 is before BG001110.
Note: This is quite a tricky exercise. If you are absolutely stuck, then at least order the accessions by the number of duplicates and hand in. - In the tab-separated files slinger.txt and hoist.txt are two columns with an accession number and a numeric result; a probability between 0 and 1. The numbers are from running 2 different programs (slinger and hoist, if you are in doubt). You must combine these probabilities - basically taking the average of the two numbers - for each accession number and write the result in a file combined.txt. The file should look like the sources, i.e. tab-separated with accession in column 1 and number in column 2. Unfortunately, the two programs have not been run from the same set of accession numbers, so some of the results are only available in one of the input files. In such case you ignore/discard the data for that accession. Only save results in the output file when the accession is in both of the input files.
- Using above method gives you too little data. You try this time to combine your two input sets differently. If an accession is in both input files you use the average, if it is in only one, you just use the number straight in the output file. This is effectively making a union of the input instead of an intersection.
- In the files geneA.txt, geneB.txt, all the way down to geneE.txt you have normalized mRNA expression data taken at the time of discovery of colon cancer for a number of patients and their survival. This is basically 2 columns in each file; The mRNA expression (x) and the number of months (y) the patient survived. For each gene you have to make a simple linear regression analysis and find 3 numbers; the α (the intercept - where the line cuts the Y-axis) and β (the slope) coefficient that describes the line running through the data points best, and the correlation coefficient (r) which describes the fitness of the line. You must identify the gene that best indicates how long the patient survives. For every gene you start calculating these values.
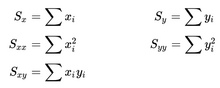 n = number of observations.
n = number of observations.
From the values you can compute the required parameters.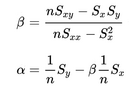
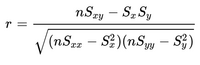
Remember to say which gene best describes survival - and why. A survival prediction can be made by calculating β * x + α, given x which is the mRNA expression.
Note: The genes will in reality interact with each other in ways that totally destroys our basic assumption for making a linear regression: That the data (gene expressions) are independent.
Make your code in a general way - there can for example be more data files. Make it easy to add them.
The gene with the best correlation coefficient is geneD, with a CC of 83.75%. - Repeat the previous exercise again with a new type of data file gene_combined.txt, which is more typical in real life. All genes are in one tab separated file. There are 3 columns; gene name, normalized mRNA expression and survival in months. There is no particular order in which the data appears and data lines for several genes might be mixed within each other.
Again, make general code. There can be more or fewer genes, and you do not need to know there names beforehand.



Exercises for extra practice
- The geneA-E.txt files all have the same structure on each line; first number is a float between 0 and 1, second number is an integer (representing months of survival after discovery of the cancer). For all files (the combined data set) find the average of the float, given the integer and display in ascending order of the integer. You need to add all the floats for a given integer together and divide by the number of floats for the integer, then you have the average for the integer. To succeed at this, you must use two dicts where the integer is the key in both. The corresponding values are the sum of the floats (for that key) and the number of times the key has been encountered in the files. Hint: Unfortunately, this does not make much biological sense, but is more in the nature of a programming exercise.
- This exercise requires exercise 6 from Simple pattern matching. Modify the code a bit so you only compute what you have to. In the data1-4.gb files count how many times the different codons in the coding sequence occurs. Display.
- This exercise builds on exercise 2 for this lesson. You must read the dna7.fsa file and translate the DNA sequences to protein sequence. Report the frequencies of the various amino acids for the entire file - all sequences (not individual sequences). That is - count how many there is of each amino acid (a total) in the translated sequences, compute the frequency of each (Number_of_this_amino_acid/Total_number_of_amino_acids) and print the results as "S 0.0123", i.e. 4 digits after the dot. You do not need to save the fasta file.